MHub / GC - Add PICAI baseline model/algorithm #60
Conversation
UpdateThis model currently works for DICOM, but the input comes from three different MRI acquisitions. For now, the pipeline is configured using a FileStructureImporter and expects the data in three different folders. This will require some data preparation on the part of the user. However, I think this should be preferred over the alternative of trying to read the DICOM metadata, since the |
|
Yes, I agree with @silvandeleemput on this one. In the general case, it may be difficult to have a reliable way to identify individual acquisitions for prostate MRI, and this should be done prior to processing with mhub. It makes sense to support this for passing inputs to mhub models. |
|
@silvandeleemput can you please point to the specific sample from IDC that can be used to confirm the function of this model? |
Yes, I will prepare and test an example of this next week on Monday. I will do this after I change the interface. It will probably be an example from one of these IDC datasets:
|
You can use any case from the Prostate-MRI-US-Biopsy images:
The corresponding DICOM data inside the Prostate-MRI-US-Biopsy can be identified using the SeriesDescription (0008,103e) tag and look something like this (can vary per case): |
|
Can you please test with a specific sample from that collection, and once you confirm it works as expected, point us to the sample that should be used for testing. |
I did, and it works as expected: |
|
@silvandeleemput I now deployed automation checks to all PR's (they will trigger once a new commit is added or we can close/open the PR). The checks currently fail as Dockerfiles now must have the In addition, I created a new build-utility which simplifies the sparse checkout. I suggest adding the following lines to all Dockerfiles, replacing the former sparse checkout for MHub models: # Import the MHub model definiton
ARG MHUB_MODELS_REPO
RUN buildutils/import_mhub_model.sh gc_picai_baseline ${MHUB_MODELS_REPO}Where E.g., during build you command would look something like this (note the repo is passed as |
I do not see this link included in the model metadata. I think it would be very valuable to have it included somewhere - this would establish direct connection between GC and MHub. |
|
@silvandeleemput do you know or can you ask - how is the repo you use for this model https://github.com/DIAGNijmegen/picai_nnunet_semi_supervised_gc_algorithm related to the earlier (published, but now archived) repo here: https://github.com/DIAGNijmegen/prostateMR_3D-CAD-csPCa. Also, can you tag Henkjan on this issue? He mentioned he uses GitHub - he might be a better person to ask this question. |
That's great, it will be a great help!
This seems like a nice feature to have, but I find it troublesome that this feature comes after I already have iterated on the established quality checklist from two months ago without being aware that such a feature was in the make. I suggest you replace these parts while you test and integrate the models. |
Agreed, I'll add it. |
|
@silvandeleemput I now deployed automation checks to all PR's (they will trigger once a new commit is added or we can close/open the PR). The checks currently fail as Dockerfiles now must have the FROM mhubai/base:latest command as the very first line.
|
Both algorithms are prostate cancer detection algorithms, but they are different: https://github.com/DIAGNijmegen/prostateMR_3D-CAD-csPCa is the 3D U-Net architecture Anindo Saha developed for this paper. It's a U-Net with attention mechanisms. https://github.com/DIAGNijmegen/picai_nnunet_semi_supervised_gc_algorithm is one of the PI-CAI baseline algorithms, which used semi-supervised learning. It's a mostly standard nnU-Net with a different loss function. It includes the trained model weights on the PI-CAI Public Training and Development dataset. |
MHub is making progress. This feature was only recently added to solve an important problem (importing from a non-existent source). As this improves readability and drastically simplifies testing of unpublished models, it is required for all models. Let's stay reasonable, this change is minor and only adds two lines to each Dockerfile. If this is a huge workload for you, please bring this up at our next meeting so we can find a solution.
Correct, you can find that this is consistent with our documentation from two months ago: |
It is indeed not that much work and I understand the nature of software in development, but don't forget that the work for each of your requests gets multiplied by ten for each open model. Also, the contract ended quite a while ago, and I have been lenient enough to provide aftercare for the open model/algorithms. For this, I asked for a quality checklist two months ago and delivered on that promise. At some point, I cannot reasonably be expected to keep adding new features unless we come to some form of agreement.
I am fine with adding this and the previously mentioned features. But as I mentioned above we have to draw a boundary somewhere. |
|
I updated the Dockerfile, and the checks have passed you should be able to continue testing. |
I did not. But at the same time, it took me no more than 5 minutes to update all the eight models we currently have. Considering that I wrote a detailed description of how to update the Dockerfile, it's a simple "replace this by that" task, that makes the testing of all the remaining 9 models from the original contract drastically faster and easier, I found it reasonable.
Wasn't integrating the models into MHub part of the contract? We're at a 0.5/10 here.
If this is an actual problem on your side, I suggest we discuss this privately in our next meeting with the NIH. |
|
@silvandeleemput Can you also provide us with three images to be shown on the algorithm page? |
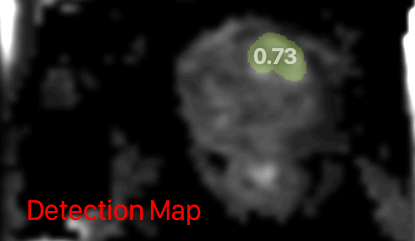
Providing images is a bit difficult; the created output image is not a true heatmap, but rather a binarized map of most-confident lesions (see https://pypi.org/project/report-guided-annotation/0.2.7/). Resulting in something like this: The images from our viewer are not the nicest (as you can tell from the screenshot above) for this particular image type, so creating nicer images is something we would prefer not to spend time on at the moment. Unless you find images like the one above acceptable. Also because the main output of the model is the prostate cancer likelihood.
|

…nd updated descriptions
|
Thank you for the feedback @miriam-groeneveld. The input / output are indeed static atm (I've an dynamic implementation prepared that I'll push soon. This will deserve some major update to a) display input and output files of the default pipeline b) allow the same for alternative workflows c) allow insights into the available data such as a likelihood value. But that's for the future). For the images, I know that non-segmentation models have a disadvantage here. However, I still think images are very valuable not only to give a good first impression and to make the model page look less technical but also to memorize model pages. Therefore, if you have some other images, maybe one you designed for the challenge, that'd be an option too. I'd also include the one you suggested. Maybe just add a red number annotation to the label (e.g., 0.74 on the green area) to indicate the nature of the area-based-probability-scores. The quality is indeed not the best but it gives the user an idea of what to expect from the model. I made up the following for now (please let me know if you do NOT agree with the usage of any of these images): |

|
What is the difference between "binarized map" and "segmentation"? How is "binarized map" not a segmentation? I understand that you do not assign 100% confidence, but it does not change the semantics that the result of the model is binary segmentation of the most probable region. Also, if there is probability threshold used for binarization, it makes sense to provide it as output of the model. Is it provided? |
|
The model outputs a detection map, which is described here: a 3D detection map of non-overlapping, non-connected csPCa lesions (with the same dimensions and resolution as the T2W image). For each predicted lesion, all voxels must comprise a single floating point value between 0-1, representing that lesion’s likelihood of harboring csPCa. It's NOT a binarized map, but rather assigns a uniform likelihood to specific regions. I think there was some confusion caused by the difference between a "true heatmap" and a "segmentation". The produced detection map can be described as a segmentation, but not as a heatmap. There is not a threshold for binarization. The voxel-level heatmap (softmax output) is transformed into the detection map using a dynamic, iterative process. This process is described in the associated paper (https://pubs.rsna.org/doi/10.1148/ryai.230031). Regarding the images, you can use the first (PI-CAI logo) as model example. The second image is taken from another paper that hasn't been referenced on the model card and was created for an algorithm that's not used here either. We can ask the author (Anindo Saha), but I don't think it makes sense for this algorithm. The third image would make sense. Can I ask where the case that's displayed is coming from? |
|
So to make sure I understand correctly. The output is probabilistic map, and the discussion above in the screenshot below was in the context of creating a picture for the model page by thresholding that map and rendering the binarized map manually, right?
If that is the case, I suggest we show the heatmap overlay for the purposes of model page. I do not think it is a good idea to show on the picture something that is not generated by the model. |

|
Also, for the model page, I suggest you show model results as overlay over T2 modality - not DWI or ADC. This will make the results a lot more appealing. And it has more relevance clinically anyway. |
|
I hope you all had a great start to the new year!! I like what @fedorov suggested. @silvandeleemput do you have such images at hand or is that doable within a minor or reasonable time investment? It would definitely improve the appearance of the algorithm's side a lot. On a separate note @silvandeleemput, did you set-up a backreference on the GC page of the challenge / algorithm? |
Thank you. I hope you had a great start as well.
I agree that it would improve the appearance of the algorithm. However, it is currently not one of the outputs of the algorithm and it would require changing the code base to output the heatmap as an option (which is a bit of work). As it cannot be generated by the algorithm at the moment, presenting such a heatmap image to the user might be confusing as it will differ from the current output.
That seems like a good idea, I'll ask @joeranbosma here (as he is the algorithm owner) to add a reference. Would a link to this page be sufficient: https://mhub.ai/models/gc_picai_baseline ? |
Never mind then - I was confused by the earlier comment from @joeranbosma #60 (comment). |
Sure, that's perfect! |
|
We're still waiting for this to be completed. Technically, we shouldn't have proceeded; please resolve this requested change ASAP so we can merge this PR and build the project. |
|
Hi all! I can add a backreference to the model on MHub (https://mhub.ai/models/gc_picai_baseline), I'll do that once the pending updates are complete. Regarding the visualization of the model output, it's a bit tricky as it's neither a heatmap, nor a binary output map, but somewhat in between. My earlier comment (#60 (comment)) describes this in detail. The third image represents the output of the model correctly. The second image is from another publication and incorrect for this algorithm and should be replaced. The prediction can also be shown as an overlay on the T2-weighted image (as suggested by @fedorov in #60 (comment)). The T2-image provides more anatomical detail compared to the ADC map. However, I would not ascribe it more diagnostic value. For cancers in the peripheral zone (accounting for about 70% of prostate cancers), the diffusion (ADC) scan is the leading modality according to PI-RADS. Since the T2-scan is also integral for the diagnosis, it's completely fine to choose it for visualization. The model weight license is not yet correct, this should be "CC-BY-NC-SA-4.0" instead of "CC-BY-NC-4.0" (check out the corresponding license file: https://github.com/DIAGNijmegen/picai_nnunet_semi_supervised_gc_algorithm/blob/master/results/LICENSE) I don't have premade visualizations of this model or detection maps, unfortunately. However, the algorithm is public, so creating these figures can be done by anyone. I'm currently not able to spend the time to make pretty figures. Best, |
{kind=link}
{kind=link}
I suggested that not because of different diagnostic value, but because prostate cancer (from what I know) often needs to be evaluated in the anatomic context. I do not think ADC is usable for assessing the location of the capsule margin, or surrounding anatomic structures. And since most likely the algorithm targeted screening applications, it would be followed by targeted biopsy, and I don't think one would use ADC for planning such biopsy. Anyway, this is really the choice of the contributor of the algorithm. It was just a suggestion, and I am completely fine with any screenshot, or with no screenshot at all.
This is potentially very problematic. What are the implications of a license SA clause: "If you remix, transform, or build upon the material, you must distribute your contributions under the same license as the original."? Arguably, one could say MHub "builds upon" this model. We absolutely cannot accept a similar license applied to MHub. Further, one could argue perhaps that outputs produced by such model are covered by that clause. And then would it mean that any repository that uses those artifacts "builds upon"? The safest bet would be to not include this model into MHub. And I would not feel comfortable including any artifacts produced by a model with such license into IDC. |
|
I suggest we bring the license topic for discussion with the NCI/Leidos contract managers at the next meeting to seek their advice. |
|
From my understanding, MHub itself, meaning the mhubio framework, the SegDB database, and all the procedures, documentation, and tutorials we developed, is separate from the deployed model in question and thus doesn't build upon the model and therefore wouldn't be affected by the same license clause. However, the actual model implementation of this specific algorithm does build upon it and thus needs to be distributed under the same license, which in practice, we already do - every MHub deployment of any model runs under the license of the original model/weights. I agree with Andrey, MHub itself of course must not be bound or restricted in any sense by any of the contributed models. Also, I agree that the best is to bring this up on our next meeting and ultimatively, we need someone with a law background to look into this. |
|
That's an interesting point about the licensing. The reasoning behind the "SA" part is that one could possibly "escape" the non-commercial part by re-distributing e.g. the model weights after making some meaningless changes. I don't know if this is true, I don't have a law background myself, and I am not aware of a good precedent on this issue. If you could bring this up with the NCI/Leidos contract managers, I would be most interested to learn what comes out of that. Based on that discussion, I can bring up the same on our end. If needed, we can also check if the "SA" part can be dropped from the model weights. |
|
May I ask what is the underlying motivation for the NC clause? |
|
Of course! The NC clause for the model weights follows from the NC clause of the data this model was trained with (see https://zenodo.org/record/6624726 for the dataset and license). For the data, we chose the NC clause in conjunction with all data providers (RUMC, ZGT, UMCG) in the public dataset. In these discussions we decided the data can be made publicly available for any researcher worldwide, but is not available for commercial parties to simply take and make money with. Commercial parties can contact us to license the data for commercial use, though. |
This PR adds the PICAI baseline model (from the PI-CAI challenge) to MHub.
GitHub Repo: https://github.com/DIAGNijmegen/picai_nnunet_semi_supervised_gc_algorithm
GC page: https://grand-challenge.org/algorithms/pi-cai-baseline-nnu-net-semi-supervised/
Algorithm I/O
Caveats
The PR target ismain, but should be something likem-gc-picai-baselineIn the Dockerfile the MHub model repo integration is currently markedTODOsince it requires integration of this PR in the main branch